原发性中枢神经系统淋巴瘤的发病机制及治疗方式
来源:医博士 | 2019-05-16 编辑:小米
原发性中枢系统淋巴瘤(PCNSL)是一种罕见的侵袭性非霍奇金淋巴瘤(NHL),常局限于大脑,眼睛,脊髓或软脑膜,而全身无受累。PCNSL总体预后,诊断和管理与其他类型的NHL不同。本研究目的在于探讨PCNSL的治疗方式以及分子受体传导机制,寻求一种更理想的治疗方法。
分子发病机制
大约90%的PCNSL病例是弥漫性大B细胞淋巴瘤(DLBCL),其余为T细胞,伯基特氏淋巴瘤,淋巴细胞和低度恶性淋巴瘤组成。PCNSL被认为DLBCL的独特亚型,并且广泛表达B细胞抗原(CD19,CD20和CD79a)。黑色素瘤相关抗原1(MUM1)和干扰素调节因子4(IRF4)阳性,CLL和BCL-6在50%的病例中表达,CD10仅在极少数情况下表达。这表明大多数PCNSL最接近于活化的B细胞(ABC)免疫表型(CD10-,BCL-6+,MUM1/IRF4+),BCL-6,CD10,BCL-2,MUM1/IRF4和ki-67对预后有重要提示作用。
诊断评估
PCNSL患者最常见的临床表现为非特异性神经认知缺陷,很少发生局灶性神经系统症状和体征。大多数免疫功能正常的PCNSL患者存在单个脑肿块,20-40%的免疫功能正常的患者仅报告了多个脑肿块,在幕上和脑室周围位置多见,如图1。头CT扫描发现病变为等密度,而磁共振成像(MRI)对PCNSL的诊断最为敏感,非增强性病变很少见,而病灶周围血管源性水肿很常见。T2加权MRI的低信号和扩散加权成像(DWI)上的扩散受限是PCNSL的典型特征。这些影像学特征可能有助于区分PCNSL和其他疾病,如感染,肿瘤性脱髓鞘病变或胶质瘤。对皮质类固醇的放射学反应很常见,但这并不能确保PCNSL的诊断,因为炎症或脱髓鞘病症也表现如此,在极少数情况下,PCNSL可能存在颅神经或神经根或局灶性脑膜增强的微小局灶性异常。
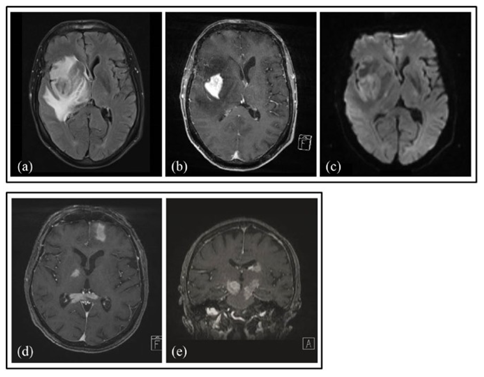
图1 两名患有原发性中枢神经系统淋巴瘤(PCNSL)的患者(a-c,d和e)的脑磁共振成像(MRI):( a)轴向液体衰减反转恢复(FLAIR)序列,显示由高信号水肿包围的低信号病变; (b)轴向,T1加权,对比度增强序列,具有强烈的均匀对比度增强; (c)扩散加权轴向序列,显示病变中的明亮信号,表明扩散受限。(d,e)轴向和冠状T1加权,对比增强序列揭示多焦点对比增强的PCNSL病变。
预后模型
对于PCNSL,已经建立了两种预后评分模型。年龄小于50岁的患者预后最好,中位成存期为8年。第一组患者中位生存期为5年,第二组患者中位生存期降至3年。年龄超过60岁,血清LDH水平升高,CSF蛋白浓度升高,并涉及大脑的深部区域。这些不利因素的0-1,2-3,4-5年的生存率为80%,48%,15%。最近的研究表明,PCNSL患者的淋巴细胞(绝对淋巴细胞计数≤875x106/L)有较低的5年生存率,该研究表明淋巴细胞减少是无进展生存期(PFS)和OS差的重要预后因素。
对于这种罕见的侵袭性中枢神经系统淋巴瘤患者的生存率,PCNSL的治疗已经有了显著的进展,但是患者复发的现象很常见,患者长期存活率依然很低。虽然尚未确定最佳的治疗方法,但目前大剂量的甲氨蝶呤仍然是PCNSL标准治疗方法。而随着PCNSL分子机制的研究进展,包含靶向药物的个性化治疗具有重大前景。
医博士编译自:Löw S, Han CH & Batchelor TT. Primary central nervous system lymphoma. Therapeutic Advances in Neurological Disorders. 2018; 11:1-16. doi:10.1177/1756286418793562.
来源:https://pubmed.ncbi.nlm.nih.gov/30305848/
声明: 所有注明“来源:医博士”的文字、图片和音视频资料,版权均属于医博士所有,转载须注明“来源:医博士”;所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。