成人ROHHAD综合征谱:一种可能的新变体
来源:医博士 | 2024-04-17
快速发作性肥胖并下丘脑调节失调、低通气和自主神经调节失调(ROHHAD综合征)是一种罕见的疾病,病因不明,出现在儿童早期,典型发病时间为1.5-7岁。到目前为止,仅报告了120例病例。ROHHAD综合征开始于先前健康的儿童贪食,导致快速发病的肥胖(6-12个月10-15公斤)。在接下来的几年中,患者出现不同程度的下丘脑功能障碍,最常见的是电解质失衡、高泌乳素血症、甲状腺功能减退和青春期开始的改变,以及自主神经障碍:包括严重的心动过缓、疼痛和温度感觉改变或过度出汗。瞳孔反应异常和斜视以及行为障碍也很常见。这些患者出现早发阻塞性睡眠呼吸暂停(OSA),随后出现中枢性低通气,导致需要家庭机械通气,并对预后产生最相关的影响,经常发生心肺骤停事件。在生命的第三个十年中没有存活的报告,到目前为止,没有报告有老年人诊断该病。本病例为第一次,一个成年人与ROHHAD综合征的诊断标准一致的情况。
病例介绍
患者女性,57岁,因食欲过盛住院,体重迅速显著增加(3个月内增加30kg,初始体重指数(身体质量指数)评估为29.7 kg·m2)、嗜睡、乏力、与疲劳相关的晕厥或晕厥前期状态、步态不稳,水平复视和外斜视相关的晕厥前事件。相关病史包括慢性自身免疫性肝炎、围绝经期和非活动性多结节性痛风。怀孕史妊娠四次,从事专业护理工作。该患者在童年和青年时期体重正常(身体质量指数为21.929.7 kg·m2 35岁时)。没有行为、精神或心理疾病,也没有服用可能导致食欲过盛的药物。
查体时,患者显示肩胛带肌肉轻度运动障碍。补充检查包括颅脑磁共振成像(MRI)显示非特异性T1下丘脑高信号,以及脊柱MRI和胸腹CT,两者均无相关异常,特别是无恶性肿瘤迹象。脑电图发现非特异性弥散性减慢活动,仅在睡眠时激活零星和短暂的双颞刺激活动。包括肿瘤标志物和免疫球蛋白在内的常规实验室检查没有显示异常。
实验室检测自身免疫性疾病显示抗核抗体HEp-2(1:640)、抗双链DNA和低补体血症呈阳性。水痘-带状疱疹病毒(VZV)的血清免疫球蛋白M和G呈阳性,除此之外,血清反应均为阴性。评估神经元、抗神经元表面、抗肌肉和神经节苷脂抗体以及卟啉研究的特定试验也呈阴性;脑脊液(CSF)模式也是非特异性的,无寡克隆条带,VZV的培养、血清学和PCR均为阴性。
出院后1个月的门诊随访期间,摄食过度和日益肥胖持续存在,并伴有间歇性白天嗜睡。最初的睡眠评估与严重的OSA一致,尽管没有进行二氧化碳监测,但没有证据表明通气不足。呼吸暂停–低通气指数为每小时54,总睡眠时间血氧饱和度<90%(t90)为11.4%,无中枢性事件,导致持续气道正压通气(CPAP)启动并充分坚持。5个月后,在重度嗜睡(Epworth评分24)的情况下,用CPAP进行滴定多导睡眠图显示,中枢呼吸暂停指数为2.1/h,占16.3%,睡眠潜伏期为1分钟,快速眼动潜伏期延长。在进行正常多次睡眠潜伏期研究、人类白细胞抗原基因分型和莫达非尼无临床反应后,放弃治疗嗜睡症。
首次入院6个月后,患者因晕厥再次住院。在急性严重低通气和对无创通气(NIV)无反应后,对其插管。入院时的初始动脉血气显示急性慢性低通气(pH 7.32,动脉氧压34 mmHg,动脉二氧化碳压(PaCO2) 62.3 mmHg,碳酸氢盐(HCO3−)30 mEq·L−1)。12周后出院,困难脱机后,患者需要延长NIV(保证容量模式的压力控制通气),每日支持时间为50%,持续反复发作的白天嗜睡持续数分钟,伴有极度氧饱和度,无呼吸困难和发作性心动过缓。内分泌评估发现中枢性甲状腺功能减退症、中枢性肾上腺功能不全、高泌乳素血症和低促性腺激素性性腺功能减退症(初步实验室结果见表1)。开始用氢化可的松治疗(每天40mg−1)和左甲状腺素(1 μg·kg−1·day−1)治疗。静脉注射免疫球蛋白(4 g·kg−1·day−1)两次给药在不同阶段产生的反应可以忽略不计。此时,肌电图显示轻度近端肌病,嗅鼻吸气压力测试正常(83 cmH2o)和对照超高场脑MRI保持不变。精神评估发现神经认知障碍,缺乏冲动控制。还进行了全面的心脏检查,包括心律分析,仅发现进行性基底性心动过缓,无其他变化。
随后的门诊随访显示进行性尿失禁,但内分泌状况保持稳定,有持续的通气支持和轻度至中度日间高碳酸血症(PaCO2 46–57 mmHg and HCO3− 26-30.8 mEq·L−1)。全面罩和口鼻罩交替使用,白天固定时间通气,以达到正常碳酸血症或允许的轻度高碳酸血症,不干扰家庭活动,基于缺乏配合和突发嗜睡和氧饱和度降低的不可预测性,排除了口腔通气。总增重为47kg(身体质量指数为45.4kg·m−2)。分子研究通过寻找PHOX2B和具有阴性结果的细胞表面神经抗原(血清/脑脊液)的其他抗体,包括N甲基d-天冬氨酸受体、α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体、γ-氨基丁酸受体A和B、代谢型谷氨酸受体1和5、LGI1、CASPR2、DPPX、neurexin-3α和IgLON5。对照腹部CT和嗜铬粒蛋白A显示非特异性结果。在正常的膈神经传导研究后,由于解剖限制,没有实施膈肌起搏。患者的行为排除了气管造口术和家庭有创机械通气。在症状出现32个月后即NIV 23个月后,在家中猝死。
Fishman等人于1965年报道了一例3.5岁男性患者,该患者具有该综合征的典型表型,出现迟发性中枢性低通气(LO-CHS),并首次描述了下丘脑功能障碍。在2000年Katz等人在LO-CHS和伴有下丘脑功能障碍的迟发性中枢性低通气综合征之间建立了独特的条件,回顾了11例表现出下丘脑功能衰竭、摄食过度、热调节障碍、嗜睡、内分泌疾病和行为不稳定特征的病例。2007年Ize-Ludlow等人创造了术语ROHHAD并建立了诊断标准。当PHOX2B基因未发现突变时,该综合征可与先天性中枢性低通气综合征(CCHS)区分。首字母缩写于2008年变更为ROHHAD(NET),包括神经内分泌肿瘤的风险。在对43例病例的广泛回顾中,Harvengt等人发现神经内分泌肿瘤的发病率为56%,其中70%的肿瘤是在最初体重增加后2年内诊断出来的。在此基础上,进行了后续CT扫描和神经内分泌肿瘤标志物检查,但没有相关发现。在同一系列中,在中位年龄4岁时检测到OSA,在5.3岁时出现中枢性通气不足,83%的患者在肥胖开始后的前5年内被诊断出OSA。24例进行了气管造口术或NIV(中位年龄4.8岁对抗6.3年)。报告的死亡率很高,在诊断后的头几年内达到60%。
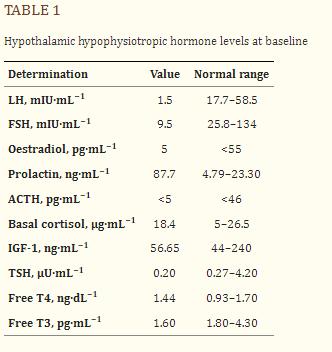
表一、下丘脑促垂体激素基线水平
讨论
与儿科患者类似,本文作者发现患者首次出现OSA并转变为主要的中心肺泡有低通气现象,这对于肺病学家来说是一项具有挑战性的评估,并且在没有气管造口术指征的患者中难以维持长时间的NIV,不影响白天的有限活动,包括行走。最初的身体质量指数和其他临床特征以及疾病的快速进展与肥胖低通气综合征不一致。作者们还认为,围绝经背景下的生理转变,包括体重和通气驱动的变化,不会对疾病全身性表现产生显著影响。与其他中央肺泡低通气综合征一样,在本病例中,可以估计高度可变和不可预测的通气需求以及触发呼吸机的潜在限制;因此,采用了特定的通风策略。
罗哈德综合征没有明确的病因,但表观遗传疾病或自身免疫过程都被认为是病因假说。然而,遗传易感性缺乏一致的结果。就像其他候选基因一样,PHOX2B突变是不存在的,比如ASCL1, 脑源性神经营养因子和HCRT。免疫球蛋白可对下丘脑功能障碍的某些表现产生部分反应,并且研究者描述了脑脊液分析后鞘内寡克隆条带的合成,免疫细胞渗入大脑,以及中脑导水管周围灰质和下丘脑局灶性炎症的MRI征象。最近研究证实了9名肿瘤相关ROHHAD患儿中有7名存在ZSCAN1(锌指蛋白和扫描结构域蛋白1)自身抗体。在本病例中,免疫介导的脑炎的完整检测结果为阴性,免疫球蛋白未见明显结果。
新生突变或常染色体显性遗传疾病的可能性在上一代中没有表达,可以解释这种散发性表现。具有典型年龄和性别差异的自身免疫性下丘脑炎的变异伴中枢性换气不足也可能与该病例有关。然而,孤立的自身免疫性下丘脑炎是罕见的,鞍上肿块存在是主要的诊断标准。与自身免疫性疾病和自主神经障碍相关的病毒前驱症状也无法确定。作者认为实验室发现与自身免疫性肝炎有关,在脑脊液和MRI分析后,VZV不能单独引发这一过程。有趣的是,新的抗下丘脑抗体和其他诊断标记物,可能应用于成人ROHHAD的频谱。
ROHHAD的表现与其他疾病重叠,包括CCHS和Prader-Willi综合征,其表型特征使我们能够排除后者。未进行全外显子组测序。另一个限制是,尽管在不同阶段发现了一些损伤的特征,但由于患者的病情,缺乏具体的测试来完成自主神经障碍的评估。
医博士编译自:Ortega-González Á, Perea-Rozas R, et al. ROHHAD syndrome spectrum in an adult: a possible new variant. ERJ Open Research. 2024; 10(1):00583-2023. doi:10.1183/23120541.00583-2023.
声明: 所有注明“来源:医博士”的文字、图片和音视频资料,版权均属于医博士所有,转载须注明“来源:医博士”;所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。