罕见遗传性疾病:HDR综合征合并肾病综合征
来源:医博士 | 2025-01-08
HDR综合征(OMIM 146255)是一种罕见的常染色体显性遗传疾病,由位于染色体10p14上的GATA3基因变异引起。GATA-3在甲状旁腺、内耳和肾脏的胚胎发育中起着至关重要的作用。HDR综合征的表型由甲状旁腺功能减退症(H)、耳聋(D)和肾脏疾病(R)组成。HDR综合征以前在中国和其他亚洲国家也有报道。本文报告了一名患有HDR综合征的中国男孩,患有早发性肾病综合征。
病例介绍
一名9个月大的男孩因腹泻住院。通过3天的常规检测,患者检测到蛋白尿。父母否认有水肿、少尿、发烧或尿液颜色异常。当地医院的常规尿检显示蛋白质3+和红细胞5-10/HP。这个男孩出生在一个非近亲结婚的健康父母家庭。在妊娠35周+4天时因胎盘早剥而剖腹产。从妊娠6个月开始检测到宫内生长迟缓。出生体重为1.47公斤(<P3th),身长为39厘米(<P3th),头围为28厘米(<P3 th)。运动发育明显被推迟。在4个月大时可以抬头。9个月大时,仍无法翻身、爬行或坐下。既往无异常病史或家族史,无母体肾病家族史。
入院时,患者的体重为7.1kg(<P3th),身高为66cm(<P3th),头围为36cm(<P3 th)。能够通过视听刺激发出声音、看和听。体检未发现明显异常畸形特征。肺部、心脏和腹部检查未发现阳性体征。未观察到其他神经发育或眼科缺陷。常规实验室检查结果,包括血常规、电解质、肝肾功能、甲状腺激素、生长激素和胰岛素样生长因子1,均正常。所有自身抗体均为阴性。C3和C4水平正常,而免疫球蛋白G(IgG)和IgA水平降低。T/B淋巴细胞亚群出现一些异常。甲状旁腺激素总水平正常。常规尿检显示蛋白3+和红细胞5-10/HP。尿微量白蛋白浓度范围为4384-5981mg/L,尿蛋白与肌酐比值范围为2.96-5.87。血液和尿液代谢分析结果均正常。根据Gesell发育时间表,估计总DQ为72,适应性行为DQ为78,大运动行为DQ为65,精细运动行为DQ是66,语言行为DQ是75,个人社交行为DQ是76。纯音测听表明感音神经性耳聋。核型是46,XY。超声检查显示肾体正常(长6.2-6.6cm,宽2.4-3.1cm),皮质和髓质边界清晰。超声心动图显示房间隔缺损(5.0mm)。脑电图和颅脑磁共振成像结果正常。
在中国MyGenologics生物技术公司的遗传学实验室,使用“遗传性肾病小组”进行了下一代测序,该小组涵盖了与这种疾病密切相关的基因。结果显示,该男孩在GATA3基因外显子3中携带半合子变体c.704C>T(p.Pro235-Leu),父母都是野生型(图1)。在人类基因突变数据库或SNP数据库(包括ALFA、ExAC、GnomAD和TOPMED)中未发现该变体。PolyPhen-2、变体Taster和GERP++分析表明该变体是致病性的,而SIFT分析表明该变异可能是良性的(ACMG指南:PS2+PM2_Supporting)。
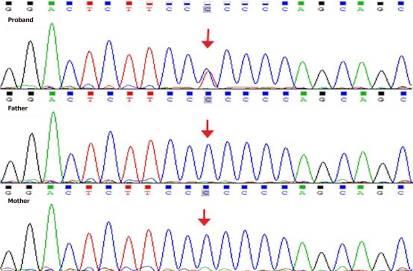
图1 GATA3基因的遗传分析。结果显示,该男孩在GATA3基因的外显子3中携带了一种新发杂合变体c.704C>T(p.Pro235-Leu)
由于存在生长迟缓、早发性肾病综合征、显微镜下血尿、感音神经性耳聋、免疫功能异常、先天性心脏病和GATA3基因中的c.704C>T(p.Pro235-Leu),诊断为HDR综合征。由于没有特定的治疗方法。患者没有接受任何特定的治疗。在3个月的随访中,蛋白尿(尿微量白蛋白:4695-5739mg/L,尿蛋白/肌酐:3.05-6.21)、显微镜下血尿(尿红细胞:5-10/HP)或生长迟缓没有明显变化。
讨论
HDR的肾脏疾病主要涉及发育异常。然而,也可以观察到肾功能异常。在这里报告的中国婴儿患有HDR综合征和肾脏疾病的病例,包括早发性肾病综合征和显微镜下血尿。
迄今为止,全世界已报告约180例HDR综合征病例。90%以上的患者患有甲状旁腺功能减退症和感音神经性耳聋,80%以上的患者出现尿路和肾脏异常。HDR综合征患者可能表现出完整的表型三联体或仅表现出一个子集。本文患者表现为生长迟缓、早发性肾病综合征、显微镜下血尿、感音神经性耳聋、T细胞免疫缺陷和先天性心脏病(房间隔缺损)。遗传分析揭示了GATA3基因外显子3中的一个新的杂合变体c.704C>T(p.Pro235-Leu)。根据临床表现和遗传结果,诊断为HDR综合征。有趣的是,患者没有出现甲状旁腺功能减退症。
在极少数情况下,HDR综合征有进一步的表现,包括女性生殖道畸形、视网膜色素变性、生长迟缓、幽门狭窄、神经异常、T细胞免疫缺陷和先天性心脏病。本例患者还表现出生长迟缓、T细胞免疫缺陷和先天性心脏病。
HDR综合征的肾脏疾病包括发育异常(如肾发育不全、发育不良、再生障碍、囊性肾病、肾盂裂孔畸形和膀胱输尿管反流)和功能异常(如蛋白尿、血尿、肾小球肾炎、近端或远端肾小管酸中毒和肾钙质沉着症)。肾病综合征和肾病水平蛋白尿在HDR综合征患者中很少见。
Chenouard等人报道了一名患有肾病综合征的HDR综合征儿童,这是一项新发现。3年后的第一次肾活检显示肾小管间质肾炎,没有发育不良或肾小球损伤,而4年后的第二次肾活检则显示节段性、弥漫性、增殖性肾小球肾炎和持续性肾钙质沉着症。Maleki等人报道了一名58岁的男性HDR综合征患者,患有慢性肾病和肾病水平蛋白尿(2.99 g/24小时)。兄弟姐妹有肾病家族史,肾活检显示局灶节段性肾小球硬化。Barakat等人报告的患者表现为类固醇耐药性肾病综合征,肾脏组织学显示胎儿样肾小球和增厚的肾小球基底膜。男孩的肾脏疾病符合早发性肾病综合征的标准,血清白蛋白浓度为24.40 g/L,尿蛋白/肌酐比值为2.96-10.86 g/g。不幸的是,该患者的肾脏病理学尚不清楚,因为父母不同意进行肾活检。
总体而言,10%的HDR患者进展为终末期肾病。HDR综合征患者发生肾功能障碍的年龄是可变的。在Chenouard等人的报告中,3岁时检测到肾功能衰竭,血清肌酐为107μmol/L,但25年后没有明显进展(血清肌酐:121μmol/L)。Maleki等人报告的患者中,慢性肾病发生在56岁,血清肌酐浓度为221μmol/L。Horta等人报告称,一名51岁的HDR综合征男性患者患有轻度肾脏疾病(血清肌酐186μmol/L),其父亲69岁死于慢性肾脏疾病。根据Joseph等人的报告,一名47岁的母亲在肾扫描中发现肾功能受损(血清肌酐:246μmol/L)和慢性实质性变化。根据Barakat等人的报告,四名兄弟姐妹在3-8岁之间死于终末期肾病。我们的患者的肾功能目前正常,随着患者年龄的增长需要随访。
小结
GATA-3在HDR综合征肾脏受累中的发病机制尚不清楚,迄今为止,还没有发现这种转录因子的潜在靶点。然而,GATA3在小鼠肾系膜细胞中表达,在系膜增生性肾小球肾炎的啮齿动物模型中显著增加,表明GATA3在正常肾小球发育中起着关键作用,可能是人类系膜细胞的有用核标志物。在过去的20年里,报告了133种GATA3变异;患者表现出很大的临床变异性,每种HDR缺陷的外显率随着年龄的增长而增加。然而,尚未确定明确的基因型和表型相关性。
医博士编译自:Ma LJ, Yang W & Zhang HW. HDR syndrome presented with nephrotic syndrome in a Chinese boy: A case report. World Journal of Clinical Cases. 2024; 12(27):6111-6116. doi:10.12998/wjcc.v12.i27.6111
声明: 所有注明“来源:医博士”的文字、图片和音视频资料,版权均属于医博士所有,转载须注明“来源:医博士”;所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。