侵袭性炎性纤维样息肉表现为小肠套叠
来源:医博士 | 2025-09-08
炎性纤维样息肉(IFP)是一种罕见的消化系统良性息肉样病变,起源于间充质组织,Vanek于1949年首次报道为“嗜酸性浸润的粘膜下肉芽肿”。IFP通常是孤立的,来自上皮组织,通常位于胃窦和小肠,较小的病变通常无症状,可在胃镜和结肠镜检查中发现。当IFP生长到一定大小时,会引起胃肠道梗阻,甚至肠套叠,表现为腹痛、腹胀、恶心、呕吐、排气停止、排便停止,甚至便血等胃肠道症状。
肠套叠是一段胃肠道伸缩到相邻段的管腔中。肠套叠在成人中很少发生,发病率为每1000000人2-3例。成人肠套叠大多为继发,因为肠腔或肠壁上的病变会导致蠕动节律紊乱,近端肠的强烈蠕动将病变与肠一起送入远端肠,常见的病理因素包括肿瘤、息肉、憩室、炎症、粘连和肠腔内异物。由于非特异性体征和症状,其诊断很困难,因此早期诊断和治疗对预后至关重要。IFP通常只涉及粘膜下层,很少突破并侵入固有肌层和浆膜下层。本文报告了2例罕见的由IFP引起的成人小肠套叠病例,IFP罕见地穿透粘膜下层。
病例1
患者81岁,中国男性,有7天的间歇性左腹痛病史,伴有恶心和呕吐。入院前两天最后一次排便和排气。腹部计算机断层扫描(CT)显示左上腹小肠梗阻,一段小肠系膜插入另一段,肠套叠的可能性很高(图1A-C)。随后,患者在手术室接受了剖腹手术,在距离Treitz韧带60厘米处发现了由2.5厘米×3.5厘米息肉样肿瘤引起的空肠段肠套叠(图1D和E)。患者接受了部分肠手术切除,手术在2小时内完成,估计失血量约为30毫升。患者恢复顺利,术后1周出院。术后对患者进行了2年的密切随访,包括随访腹部CT扫描,未发现复发迹象。
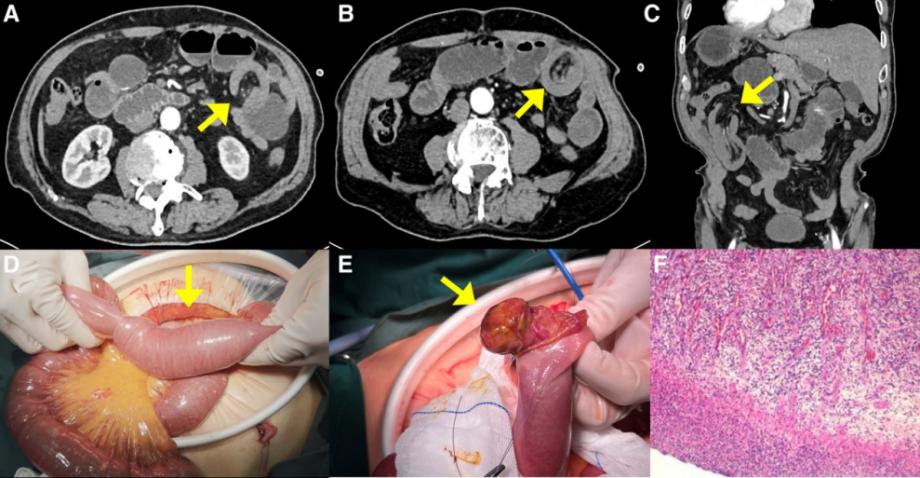
图1.:(A-F)病例1的围手术期放射学特征、术中表现和术后组织病理学特征。(A、B、C)腹部CT显示肠套叠的“靶征”和“杯状征”(黄色箭头)。(D) 手术探查显示,肠的近端部分及其肠系膜进入更远端的部分。(E) 观察到3.5 cm×2.5 cm的病变(黄色箭头)。(F) 显微镜检查显示,病变侵犯了固有肌层,并观察到含有嗜酸性粒细胞的炎性细胞背景。CT=计算机断层扫描
病理检查显示息肉样肿物侵犯了固有肌层。肠黏膜被侵蚀,下方形成肉芽组织,血管丰富,血管周围可见散在分布的粗大异形囊肿,核仁明显,可见核分裂,细胞质丰富(图1F)。免疫组织化学显示,异形细胞对ALK、Ki-67和B-Catenin呈阳性,对HMB45、CD117、CD34、SMA、Desmin、DOG1、CD31和ERG呈阴性。结合免疫组化染色,梭形细胞对CD34呈弱阳性,对Ki-67呈阳性,对STAT6、DOG-1、AE1/AE3、ALK(Ventana-D5F3)、GFAP、S-100、SOX10呈阴性。粘膜下层可见增殖的间充质细胞,呈梭形和星状,伴有嗜酸性浸润,与IFP一致。
病例2
一名49岁的中国女性,有5天的间歇性下腹胀痛病史,尤其是脐周腹痛。腹部CT显示,左下腹有一段小肠插入另一段小肠,肠内积聚液体和气体(图2A和B)。患者在紧急情况下接受了剖腹探查。在剖腹手术中,距离回盲部40cm处发现了回肠肠套叠。手动复位后,发现一个3.6×3.7cm的息肉样病变浸润到浆膜层,突出并完全闭塞管腔。患者接受了持续2.5小时的部分肠切除术,估计失血量为20毫升。恢复顺利,术后8天出院,没有任何术后并发症。术后对患者进行了1.5年的密切随访,在此期间,没有报告腹部不适。后续的腹部CT扫描也没有发现复发的证据。
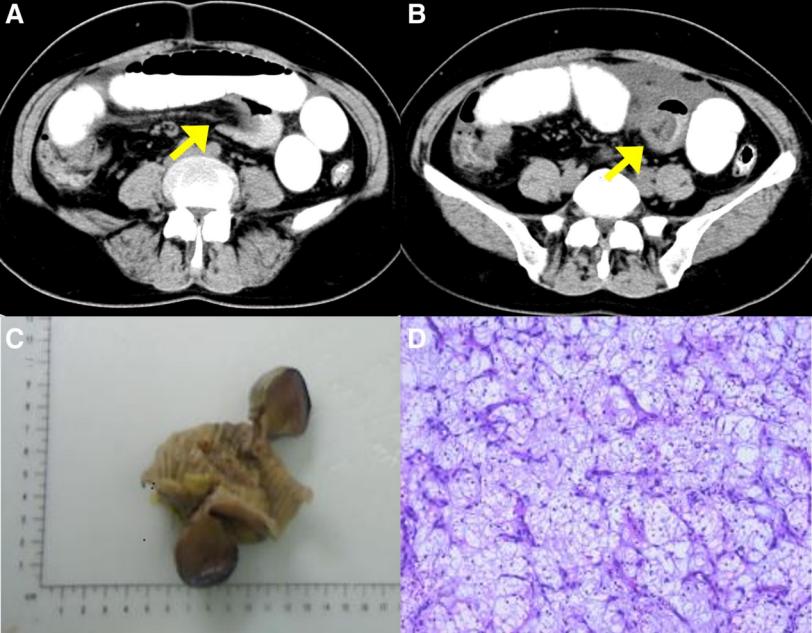
图2:(A-D)病例2的围手术期放射学特征和术后组织病理学特征。(A和B)腹部CT显示肠套叠的“靶征”和“杯状征”(黄色箭头)。(C) 大体标本:观察到3.6×3.7cm的息肉样病变,侵犯浆膜层。(D) 可见大量炎性细胞的背景,其中以淋巴细胞、浆细胞和嗜酸性粒细胞为主。CT=计算机断层扫描
病理检查显示,粘膜中存在3.6×3.7cm的息肉样病变,侵犯了浆膜层(图2C)。显微镜下,病变表现为糜烂、明显的间质水肿、粘液变性和血管增生。可见大量炎性细胞,以淋巴细胞、浆细胞和嗜酸性粒细胞为主(图2D)。免疫组织化学检查显示,CD34和PDGFRα呈阳性,Ki67呈局灶性阳性,CD117、DOG-1、SMA、S-100、ALK80、ER、PR和Desmin呈阴性。这些形态学和免疫组织化学结果与IFP相符。
讨论
IFP可以在整个胃肠道中发现,但在胃中更常见(66.7%),其次是小肠(21.1%)和结肠(8%),在其他部位很少见(<3%)。2例IFP均位于小肠,1例位于空肠,另1例位于回肠,这种情况很少见。IFP的病因和病理生理机制尚未完全确定,局部感染(蠕虫、幽门螺杆菌)、过敏反应、自身免疫过程或宿主对未知刺激的过度反应都被描述为IFP发展的可能原因。一些研究表明,IFP是一种由酪氨酸激酶受体PDGFRA激活突变驱动的良性肿瘤,通常存在具有一定家族聚集性的致癌PDGFRA突变。遗憾的是,我们没有对这2例病例进行基因突变分析。
形态学上,大多数IFP表现为从粘膜深层或粘膜下层出现的孤立、无柄或带蒂的息肉样肿块。2例IFP突破粘膜下层,1例侵犯肌肉层,另1例甚至侵犯浆膜层,无外周淋巴结肿大。据报道,IFP可以通过固有层侵入浆膜,并发生透壁生长,这是一种极其罕见的观察结果。报告表明,IFP具有潜在的侵袭性,有时伴有外周淋巴结肿大。尽管IFP在固有肌层下的传播极为罕见,但识别类似病例和进一步研究将加深我们对这种肿瘤性质的理解。一项研究发现,在50例胃IFP中,4例(8%)与同一解剖区域的并发腺癌或腺瘤有关。
小结
对文献的全面回顾揭示了2例记录在案的胃肠道内复发性IFP病例。尽管目前没有直接证据表明IFP是恶性肿瘤,但上述文献中报告的侵袭性生长模式、复发以及与腺癌或腺瘤的关联特征强烈支持IFP的恶性性质。
医博士编译自:Tang Y, Cui X, Zhao Z, et al. Rare invasive inflammatory fibroid polyp presenting as small bowel intussusception: Two case reports and review of the literature. Medicine (Baltimore). 2025; 104(13):e41956. doi:10.1097/MD.0000000000041956
声明: 所有注明“来源:医博士”的文字、图片和音视频资料,版权均属于医博士所有,转载须注明“来源:医博士”;所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。