原发性肾黏膜相关淋巴组织淋巴瘤伴免疫性血小板减少症
来源:医博士 | 2025-10-14
粘膜相关淋巴组织(MALT)淋巴瘤,也称为结外边缘区B细胞淋巴瘤,约占非霍奇金淋巴瘤的5%,主要影响胃、肺和唾液腺等器官。由于肾实质缺乏淋巴组织,原发性肾淋巴瘤很少见,故原发性肾脏MALT淋巴瘤的发生率非常低。
免疫性血小板减少症(ITP)是一种自身免疫性疾病,其特征是免疫介导的血小板破坏和血小板生成减少。继发性ITP可由免疫缺陷、病毒感染、骨髓疾病或淋巴增生性疾病引起,如慢性淋巴细胞白血病、非霍奇金淋巴瘤(NHL)和霍奇金淋巴瘤。尽管如此,ITP和NHL同时存在的情况相对罕见,ITP在NHL中的患病率估计为0.76%。对PubMed、Web of Science、Scopus和Google Scholar数据库的系统搜索显示,之前没有直接记录ITP与原发性肾MALT淋巴瘤共存的报告。
本文介绍一例罕见的原发性肾MALT淋巴瘤病例,该病例在ITP治疗后被诊断,并详细记录了其临床进展、放射学特征和临床病理特征。
病例介绍
一名60岁男性因持续头晕和疲劳而接受血液学检查。他否认体重减轻、发烧、盗汗、出血倾向和近期药物暴露。体检未发现淋巴结病或其他异常。血小板计数持续下降(<20× 10⁹/L)。 经两次单独的实验室检测证实,白细胞和血红蛋白在正常参考范围内。肝功能测试、肾功能测试(包括血清肌酐和血尿素氮)、电解质水平和凝血曲线均未显示出明显异常。综合血清学排除了感染性(HBV/HCV/HIV)、肿瘤性(LDH/β2M/PSA)和自身免疫性病因(ANA、ENA组、抗dsDNA、抗着丝粒阴性)(表1)。
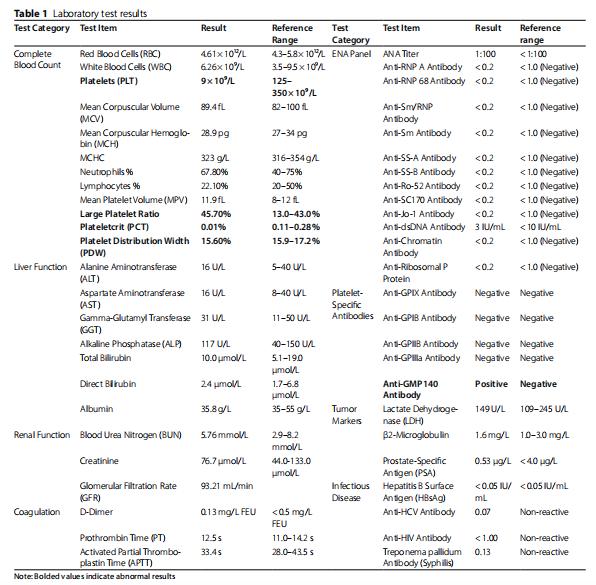
表1
血小板特异性抗体检测显示抗GMP140抗体阳性。骨髓吸出物显示红系、粒细胞和单核细胞谱系正常,同时巨核细胞成熟受损(每个低倍视野21个巨核细胞,仅包括1个血小板生成形式),没有母细胞白血病、骨髓纤维化或淋巴瘤浸润,有助于排除骨髓增生性肿瘤(MPN)和骨髓增生异常综合征(MDS)。同时进行的外周血分析证实了明显的血小板减少症,其特征是血小板稀少、分散,其他谱系没有发育不良。计算机断层扫描(CT)和CT血管造影确定了8.7×5.6×3.0 cm浸润性左肾肿块桥接肾窦和下极,表现为实质和血管浸润(图1A-D)。肾脏动态闪烁扫描显示,尽管左肾形状不规则,但两个肾脏的血流灌注和肾小球滤过功能正常(图2)。
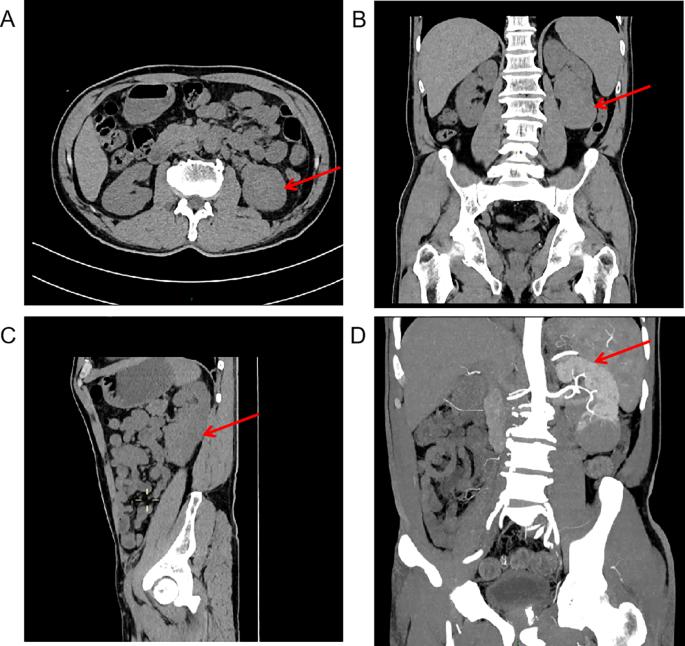
图1 肾肿瘤的计算机断层扫描和肾动脉造影。(A)腹部CT平扫显示左肾软组织肿块,箭头为红色。(B)冠状位CT平扫显示肿块位于左肾窦和左肾下极,如红色箭头所示。(C)矢状CT平扫显示,肿块位于左肾窦和左肾下极,如红色箭头所示。(D)肾动脉计算机断层扫描血管造影扫描描绘了左肾动脉与位于左肾窦和下极的软组织肿块相邻并沿其穿过,由红色箭头标记
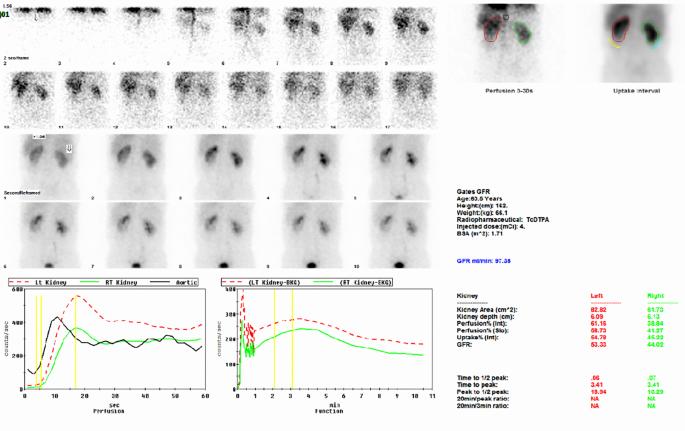
图2 患者的肾脏动态闪烁扫描结果。肾动态闪烁扫描显示,两个肾脏的血流灌注正常,左肾和右肾的肾小球滤过率分别为53.3 mL/min和44.1 mL/min
由于严重血小板减少症,开始口服糖皮质激素治疗(泼尼松)、皮下血小板生成素和血小板输注,旨在避免颅内出血等严重并发症。血小板残留 < 30 × 109/L持续7天,然后逐渐恢复(第13天恢复正常)。随后,患者被转移到泌尿科进行最终治疗。泌尿学评估发现,在连续尿检中,左肋椎压痛伴持续性白细胞尿(++/+++)和意外的粘质沙雷氏菌尿,没有任何伴随的泌尿系统症状,如排尿困难、尿频或血尿。在全面评估肿瘤可切除性和患者对活检相关并发症的担忧后,最终在没有术前活检的情况下进行了根治性肾输尿管切除术。术后恢复顺利,在六个月的随访期间,血小板计数始终保持在正常范围内。连续CT扫描证实持续缓解,没有肾淋巴瘤复发的证据。
肾输尿管切除标本的组织病理学评估显示,9.0×5.0×4.5cm中等密度的灰白色肿瘤浸润肾中下极(图3)。组织病理学评估显示,小淋巴细胞密集浸润,核轮廓圆形至轻度不规则,染色质中度分散,嗜酸性细胞质苍白,核仁不明显,累及肾门淋巴结、输尿管壁和肾周脂肪组织(图4A-D)。肿瘤细胞对CD19/CD20/CD79a/CD79b/MNDA/IRTA1/BCL2免疫表型呈阳性,但对CD3/CD5/CD10/Cyclin D1/MUM1/BCL6呈阴性(图4E-F)。免疫组织化学分析证实肿瘤细胞中没有免疫球蛋白轻链(κ/λ)表达,爱泼斯坦-巴尔病毒的原位杂交显示EBER阴性。病理学家的最终诊断是位于肾脏的结外边缘区淋巴瘤。
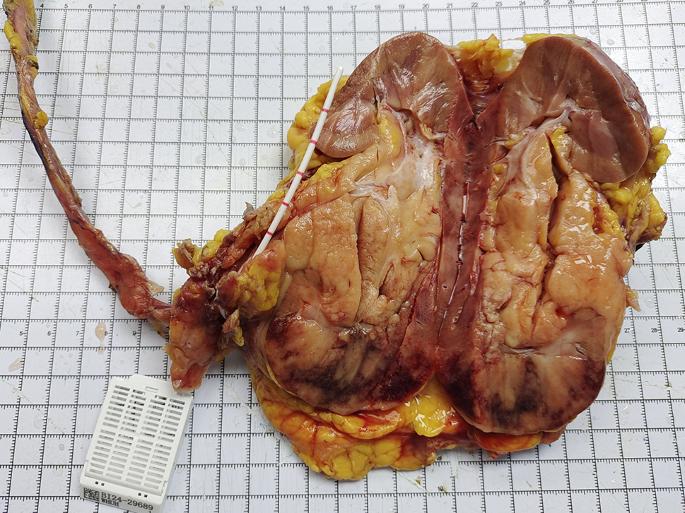
图3 原发性肾MALT淋巴瘤的大体病理。切除的标本显示,肾脏中下极有一个边界清晰的灰白色肿块,大小为9.0 × 5.0 × 4.5 cm,与常规肾细胞癌不同,其具有完整的纤维包膜和中等密度
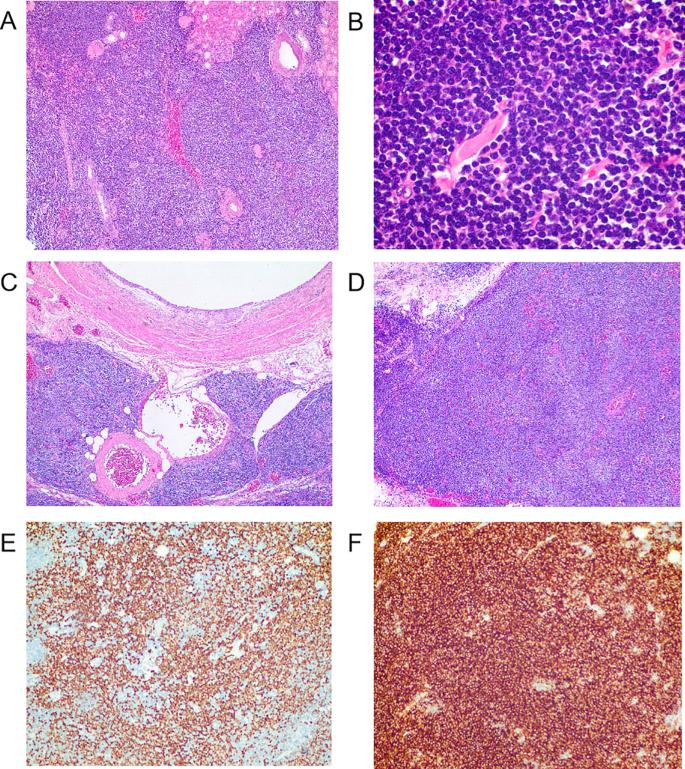
图4手术标本的组织学发现。(A)淋巴组织浸润肾实质,包括肾小球和肾小管结构。苏木精-伊红染色,原始放大倍数×40。(B)组织病理学评估显示,小淋巴细胞密集浸润,核轮廓圆形至轻度不规则,染色质中度分散,嗜酸性细胞质苍白,核仁不明显,浸润肾实质。苏木精-伊红染色,原始放大倍数×400。(C)肿瘤淋巴细胞穿透输尿管浆膜层并侵入输尿管壁。(D)肿瘤淋巴细胞浸润左肾门淋巴结。(E)免疫组织化学显示,肿瘤淋巴细胞对MNDA呈阳性。(F)免疫组织化学显示,肿瘤淋巴细胞对CD19呈阳性
MALT淋巴瘤是一种独特的惰性B细胞淋巴瘤,约占新诊断淋巴瘤病例的7-8%。虽然通常涉及结外部位(胃黏膜、眼附属器、乳腺、肺、唾液腺),但肾脏受累仍然非常罕见。原发性肾淋巴瘤(PRL)主要表现为弥漫性大B细胞淋巴瘤,很少观察到低度MALT变异。
MALT淋巴瘤的诊断基于不同的病理特征:异质性小B细胞包括小淋巴细胞、中心细胞样细胞和单核细胞样细胞,经常散布着分散的免疫母细胞和中心细胞样细胞。在免疫表型上,这些肿瘤细胞表达B细胞抗原(CD20、CD19、CD79a、PAX5和Bcl-2),但缺乏CD5/CD10。
根据提供的免疫表型特征,我们病例中的肿瘤细胞表现出CD19、CD20、CD79和Bcl-2的表达,而缺乏CD3、CD5、CD10、Bcl-6和Cyclin D1的表达。CD10和Bcl-6的缺失有效地排除了滤泡性淋巴瘤的诊断,CD5和Cyclin D1的缺失排除了套细胞淋巴瘤的诊断。因此,该肿瘤被诊断为原发性肾MALT淋巴瘤。
自身免疫性疾病(AD)患者面临的淋巴瘤风险比健康人高70倍,这可能是由于靶器官的慢性抗原刺激。ITP是一种与NHL相关的免疫血液学并发症,可在NHL临床症状出现之前、同时或随后出现。一项全国性的瑞典队列研究显示,ITP诊断后NHL发病率显著升高,与年龄匹配的普通人群对照组相比,男性的相对风险增加了7.4倍。尽管ITP通常在NHL发病前出现,但这两种疾病之间的确切因果关系仍有待最终确定。一方面,经验证据表明,肿瘤细胞具有产生抗血小板抗体或通过抗原模拟机制引发自身免疫的能力。另一方面,在自身免疫性疾病患者中观察到的免疫系统失调可能会导致肿瘤免疫力下降,从而增加发生淋巴增生性恶性肿瘤和实体瘤的可能性。几份记录淋巴瘤治疗后ITP缓解的病例报告进一步证实了这些疾病之间的潜在相互关系。Alexander W.Hauswirth等人的研究表明,NHL前的ITP对皮质类固醇和高剂量免疫球蛋白反应良好,这与我们的发现一致。在本例中,患者的ITP在术前皮质类固醇治疗后实现了持续缓解,术后未观察到复发。然而,在这种情况下,ITP和NHL之间的潜在因果关系仍未确定。总之,肿瘤、自身免疫性疾病和慢性炎症之间的相互作用非常复杂。ITP作为副肿瘤现象或对恶性细胞的错误免疫反应的确切性质需要进一步研究以全面了解。
研究表明,MALT扩散发生在高达25%的非胃肠道MALT淋巴瘤中,胃和肺MALT淋巴瘤的淋巴结受累更为常见。在我们的病例中,观察到肾门淋巴结受累和输尿管壁侵犯,但未检测到肺或其他器官的远处转移。虽然PET-CT可以提供肿瘤转移的全面评估,但目前的指南并没有特别推荐。在这种情况下,患者接受了手术切除作为唯一的治疗方法,在术后六个月的随访中没有复发的迹象。有证据表明,利妥昔单抗等治疗药物在淋巴瘤治疗期间可能会诱发严重的ITP,因此需要谨慎选择术后化疗方案。
小结
肾淋巴瘤经常被误诊为肾细胞癌,这突显了将其纳入孤立性肾肿块鉴别诊断的重要性。关键的鉴别诊断还包括血管平滑肌脂肪瘤、嗜酸细胞瘤、肾盂癌、继发性肾淋巴瘤和黄色肉芽肿性肾盂肾炎,最终诊断依赖于特征性影像学发现的多模式整合和验证性组织病理学评估。即使经过长期随访,MALT淋巴瘤患者也经常报告晚期或远处复发,在某些情况下需要终身监测。
医博士编译自:He J, Deng C, Huang Y, et al. Primary renal mucosa-associated lymphoid tissue lymphoma coexisting with immune thrombocytopenia. World Journal of Surgical Oncology. 2025; 23(1):265. doi:10.1186/s12957-025-03916-w.
声明: 所有注明“来源:医博士”的文字、图片和音视频资料,版权均属于医博士所有,转载须注明“来源:医博士”;所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。